■業界動向
1.レムデシビル、COVID-19 治療薬として特例承認-NIAID主導プラセボ対照P3で主要評価項目達成-
■業界動向
1.レムデシビル、COVID-19 治療薬として特例承認
-NIAID主導プラセボ対照P3で主要評価項目達成-
米国立アレルギー・感染症研究所(NIAID)は4月29日、新型コロナウイルス感染症(COVID-19)が進行し、肺炎を呈している入院患者を対象とした米ギリアド・サイエンシズの抗ウイルス薬レムデシビルの無作為化、二重盲検、プラセボ対照第3相ACTT試験の1063人の速報結果として、主要評価項目(回復までの期間)を達成したと発表した。
試験は2月21日に開始され、米国、欧州、アジアの68の医療機関が参画。日本からは国立国際医療研究センターが参画した。
速報結果によると、レムデシビル投与群は、プラセボ群に対して31%回復までの期間を早め、統計学的有意差を示した(p<0.001)。回復までの期間の中央値は、プラセボ群の15日に対して、レムデシビル群は11日だった。
また、死亡率はプラセボ群が11.6%であったのに対して、レムデシビル群は8.0%と統計学的有意差は示せなかった(p=0.059)ものの、ベネフィットが示唆された。
プラセボ対照の第3相試験で優越性を示した意義は大きく、今後はFDAの承認タイミングに焦点が移る。これまでFDAは、緊急時使用許可(EUA)制度の下、COVID-19治療薬としては唯一クロロキン/ヒドロキシクロロキン、COVID-19検査キットでは、5分で陽性判定できる「ID NOW」などに供給と緊急使用の許可を出してきた。
5月1日、FDAはレムデシビルにEUAを出した。NIAID主導の第3相試験の肯定的データ発表からわずか2日後のことである。EUAは、新型コロナウイルスへの感染が疑われるか、検査で感染が確定し重度の症状を呈する患者に限定。重度の症状とは、室内気下の酸素飽和度(SpO2)が94%以下であるか、酸素補給または人口呼吸器、または体外式膜型人工肺(ECMO)を必要とする場合と定義されている。
日本では、安倍晋三首相が4月27、28日の国会答弁でレムデシビルの特例承認に言及。厚労省は5月7日に特例承認した。米国でのEUAは緊急時の一時的措置であり、実質日本は世界初承認国となった。これにより厚労省は国内必要量の確保に全力を挙げる。ギリアドによると、現段階の供給量は、出荷可能な製剤と最終工程にある製造中の薬剤を合わせて150万回投与分。投与期間を10日間と想定した見積もりでは、14万人分に相当する。この150万回の投与分全てのグローバルでの無償提供を約束している。
日本において特例承認後、無償提供されたレムデシビルを保険外併用療養で使用することになるとみられる。当初、日本への供給量は限られると思われ、投与する患者選択が課題になりそうだ。
ギリアドは、20年10月までに50万人分、12月までに100万人分、必要であれば21年には数百万人分の生産量を目標としている(各患者が10日間投与を受けると想定して計算)。海外製造会社との提携も模索している。安定供給できるかどうかは将来の日本での薬価収載の条件になる。
短期間投与で済む可能性
一方、ギリアド・サイエンシズは4月29日、重度のCOVID-19患者を対象とした非盲検第3相SIMPLE試験の初期データとして、5日間投与と10日間投与によるレムデシビルの有効性は同程度だったと発表した。
チーフ・メディカル・オフィサー(CMO)であるマーダッド・パーシー氏は「NIAIDが実施しているプラセボ対照試験(ACTT試験)のデータを補完するものとなり、レムデシビルの最適な投与期間を判断する一助となる。今回の試験は、一部の患者は5日間の治療を受けられる可能性を示すもので、現時点でのレムデシビルの供給量で治療可能な患者を大幅に増やせる可能性がある」とコメントした。なお、NIAIDのACTT試験は、最長10日間投与で行われている。
第3相SIMPLE試験は、重度患者と中等度患者を対象に2月26日に開始され、当初1000人(重度:肺炎の所見を示し酸素飽和度の低下が見られるものの人工呼吸器を必要としない400人/中等度600人)を登録する計画だった。それが4000人(人工呼吸器の使用例を含む2400人/1600人)への拡大を経て、7600人(6000人/1600人)に及んでいる。
世界各地の180の医療機関が参画。日本においては4月14日から投与が開始され、ClinicalTrials.govによると、横浜市立市民病院、名古屋市立東部医療センター、東京都立墨東病院が参画している。
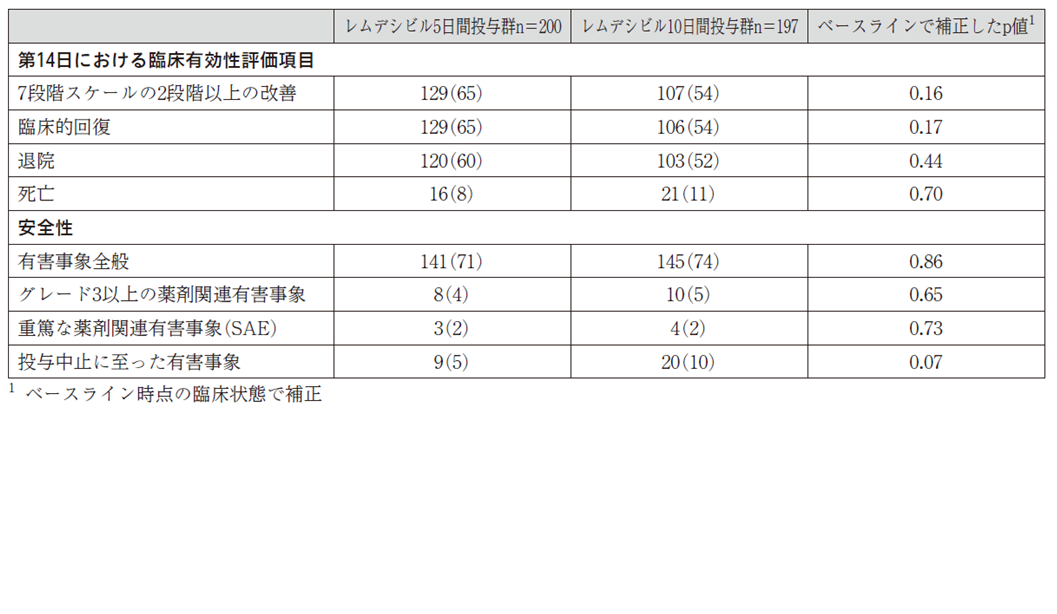
4月29日にギリアドから発表されたのは、当初の約400人の重度患者の結果(日本の患者は含まれていない)である。患者は5日間投与または10日間投与に1:1に無作為に割り付けられている(200人/197人)。比較対照群は置いていない。
臨床的改善は、試験計画書に示した7段階スケール(退院~酸素療法の必要性~死亡)でベースラインからの2段階以上の改善が見られることと定義。臨床的回復は、酸素療法と治療が不要となるか、退院に至ることとした。
表を見ると、死亡率は5日投与群で16人(8%)に対し10日投与群で21人(11%)、退院率は5日投与群で120人(60%)、10日投与群で103人(52%)―などとなっている。なお、探索的解析からは「発症後10日以内にレムデシビルの投与を受けた患者は、発症後11日以降に投与を受けた患者に比べ、転帰が良好だった」とされ、より早期に治療を開始する重要性が示唆された。
新たな安全性シグナルは認められず、忍容性はおおむね良好だったとしている。患者の10%以上に認められた有害事象は、悪心と急性呼吸不全。グレード3以上の肝酵素(ALT)上昇は患者の7.3%(n=28/385)で認められ、3.0%(n=12/397)は肝酵素上昇のため投与を中止したという。
第3相SIMPLE試験のもう一つの中等度患者を対象とした試験では、標準治療(対症療法)に加え、レムデシビル5日間および10日間投与群と、標準治療のみの群とを比較して評価している。
初期600人の結果は、5月末に得られる見込みといい、ギリアド日本法人によれば、4月14日に投与が始まった日本の患者のデータが含まれる可能性がある。
同じ29日には、中国において登録症例数不足のために早期中止となった重度患者を対象とした第3相試験結果がランセット誌で発表されており、NIAIDおよびギリアドの発表は、これにぶつけた格好となった。